The rapid progress of next-generation sequencing (NGS) technologies and third-generation single-molecule sequencing (TGS) technologies have moved bio-medical researches into big data era. The big data issue makes sequencing-based tasks, including bioinformatic analyses, data statistics & visualization, and data transfer & storage, become more challenging than ever, and inspires new ideas for bioinformatics and bio-medical researches. Especially, de novo genome assembly of human and non-model organisms for personalized medicine, and Genome breeding, will pave the way to help analyze the mechanism of diseases and decipher the secrets inside the genome.
However, few existing methods and software that can deal with the hybrid genome assembly of NGS linked-read (10x Genomics) data and TGS long-read data currently, and the results are limited and not satisfactory. However, these tools do not fully utilize the information inside the raw sequences. For emerging sequencing technologies like Hi-C (Chromosome conformation capture) and Bionano (optical genome mapping technology), both of them also impulse us to integrate all available sequencing approaches to form a more comprehensive genome than ever.
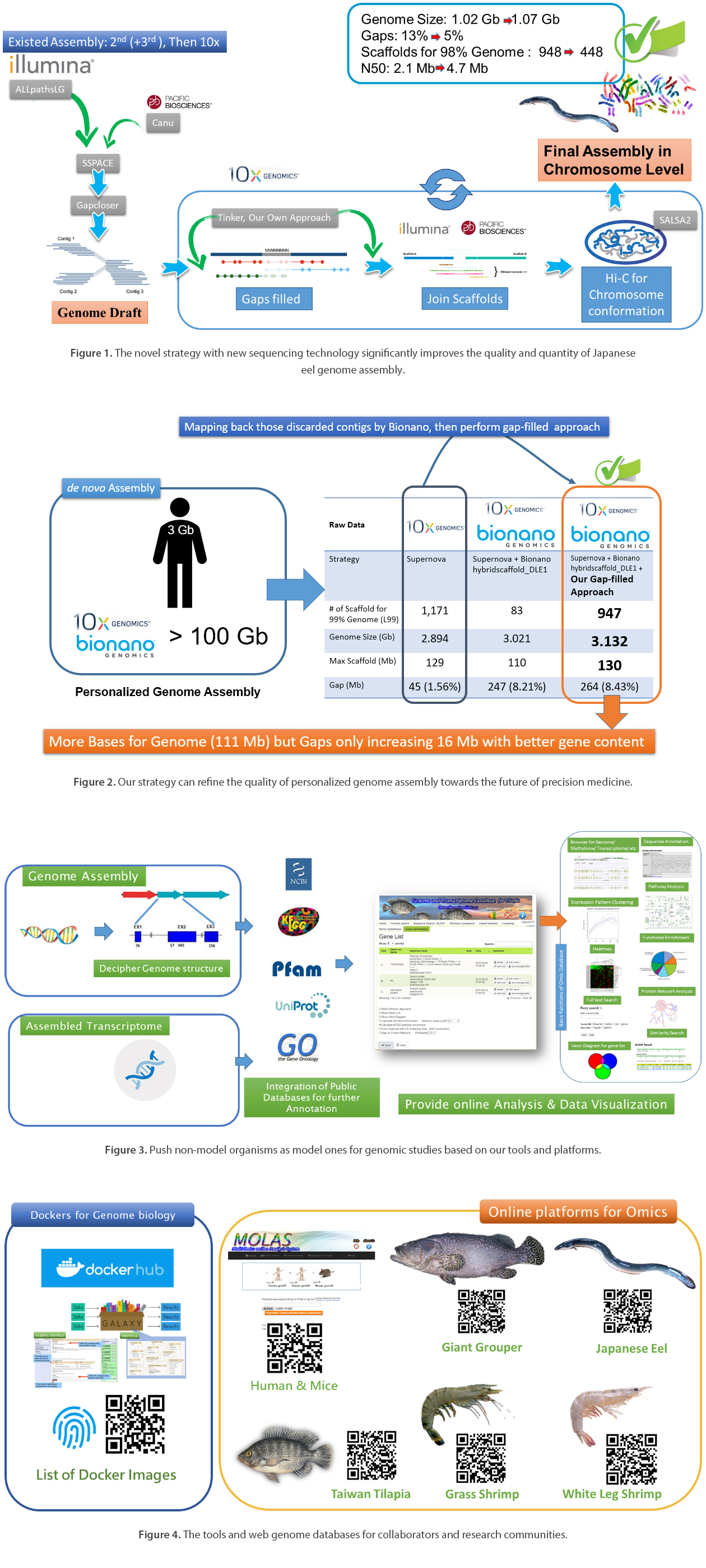
Currently, we have generated and collected many sequencing data, including NGS reads, linked reads, TGS reads, and Bionano optical mapping outputs. Meanwhile, we have started to develop a new hybrid, and spiral approach by the integration of gaps closing & scaffolding algorithms using local de novo assembling of link reads (10x genomics). It improves draft genome assembly in terms of both contiguity and completeness on aquaculture species (figure 1) (i.e., Japanese eel, Taiwan Tilapia, Giant Grouper).
Besides, our new algorithm can integrate a large number of raw reads from 10x Genomics and Bionano optical mapping to assemble de novo personalized genome draft with more genome contents and fewer gaps increasing up to chromosome-level (Figure 2). For easing the computational burdens caused by massive computing tasks of local assembling, we seek the collaboration of Taiwan Microsoft Inc. on cloud computing.
To better understand on the assembled genome, we implemented a web-based framework named as MOLAS. MOLAS can promote the genomic studies of these non-model species to the level of model organisms such as fruit flies or mouse, and to assist the research community for more in-depth analyses (Figure 3). All software and tools derived from this study are released to the public by GitHub and DOCKER images. Furthermore, several web databases are also available for the research community (Figure 4).