Principal Investigators
Postdoctoral Fellow
- Yu-Jung Chang
- Yu-Wei Tsay
Group Profile
Our current research is focused on bioinformatics for “omics” studies, classified into two main areas: (i) genomics and transcriptomics, and (ii) proteomics and metabolomics. These areas are described below.1. Genomics and Transcriptomics
Developing Methods for Sequencing Data.
With the ascension of next-generation sequencing (NGS) as the predominant technology for genomics and transcriptomics studies, we have devoted ourselves to developing new methodologies and tools for analyzing NGS data. For NGS read mapping, we have developed an ultra-efficient divideand- conquer algorithm, called Kart, which divides a read into small fragments that can be aligned independently. Our experiments show that Kart is 3 to 10-times faster than other aligners and still produces reliable alignments, even when the error rate is as high as 15%. The same strategy has also been applied to our RNA-seq mapper, and we obtained superior results to comparable technologies. For de novo genome assembly, we have proposed an extension-based assembler, called JR-Assembler. This tool can assemble giga-base-pair genomes from lllumina short reads, while achieving better overall assembly quality with faster execution times. Moreover, JR-Assembler has advantages of improved memory usage and execution time that increases slowly as the read length increases.
Integrated Tool and Platform Development for Sequencing Data.
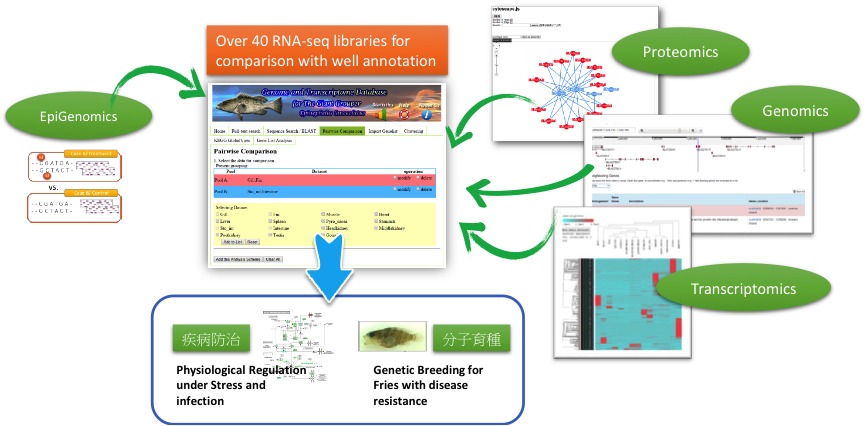
We are developing pre-assembly and post-assembly analytics for NGS and third generation sequencing (3GS), based on a MapReduce framework, which will predict repetitiveness and sequencing errors of a read and optimize efficacy and efficiency of de novo genome assembly. Meanwhile, we are also developing a cloud-based architecture to further speed up the execution of de novo genome assemblers. For assembled genomes, we have implemented pipelines that can decipher genome structure, annotate genes and rapidly estimate expression profiles. Using our own web platform (http://molas.iis.sinica.edu.tw), and our integrated approach toward genomics, transcriptomics, proteomics, methylomics and other omics, we and our collaborators are tackling projects related to fusion gene discovery from clinical biopsies, functional annotation for non-model organisms (e.g., Giant grouper, http://molas.iis.sinica.edu.tw/grouper2016 and Japanese eel, http://molas.iis.sinica.edu.tw/jpeel2016), precision phenotyping of pathogens and identification of mechanisms that restrict virus replication for vaccine development.
Transcriptional Regulation.
Transcription factor binding is determined by the presence of specific sequence motifs and chromatin accessibility. We have developed a random forest model, which considers the chromatin state and DNA structure properties, and significantly improves the accuracy of transcription factor binding site prediction. In addition, we have made two important discoveries related to nucleic acid structure/function: (a) The occurrence of non-B DNA structure motifs is significantly correlated to exon skipping events, demonstrating that structural blockage plays a role in transcriptional-coupled splicing. (b) Enhancers are highly related to DNA looping and the transcripts (eRNAs) may be associated with the selective activation of enhancer target genes in mouse.
Regulatory Networks.
To explore important nodes/hubs and fragile motifs in a complex biological network, we have implemented eleven topological algorithms as cytohubba (http://apps.cytoscape.org/apps/cytohubba). Since 2011, it has been downloaded over 9,000 times and cited over 180 times. The new version of cytohubba was released in Jan, 2017 and within three months was downloaded more than 400 times. All the algorithms will be interfaced into the Galaxy framework and distributed as an image via Docker and virtual machine (VM). In this way, these algorithms can be easily integrated into our analytic pipelines and support broad applications for the biomedical research community.
2. Proteomics and Metabolomics
Bioinformatics for Mass Spectrometry-based Proteomics.
Mass Spectrometry (MS) has become the predominant technology for proteomics research. There are two complementary approaches for MS experiments: a bottomup approach (also called shotgun) and a top-down approach. Currently, most proteomics research uses the shotgun approach. Thus, we have developed applicable computational methods and tools for protein identification and quantitation. Though many sequence database search tools are available for protein identification, they cannot be used to identify intact glycopeptides. We have proposed algorithms and implemented an automated tool, called MAGIC, for intact glycoprotein identification. Furthermore, we have developed a web server, called MAGIC-web, to tackle large-scale and targeted glycoprotein identification. For quantification of individual proteins, we are currently developing a new version of our previously published tool, Multi-Q, for isobaric-labeling quantitation analysis of TMT 10-plex-labeled samples. Top-down proteomics analysis has been receiving attention of late, because it allows for the characterization and identification of post-translational modifications. However, the data analysis for top-down proteomics is challenging. Much of the difficulty lies in deconvoluting charge state peaks, in order to detect a proteoform. We have recently proposed a method, called DYAMOND, to solve this problem and have developed a tool, called iTop-Q, which implements DYAMOND for top-down proteomics quantitation.
Bioinformatics for Glycan Synthesis and MS-based Metabolomics.
Although the synthesis of oligosaccharides is a mature methodology, the practice is still limited to specialized laboratories. To reduce the complexity of intermediate separation and protecting group manipulation, we have developed an automated method for programmable one-pot oligosaccharide synthesis. In MS-based metabolomics, we have developed an automated metabolite quantitation tool, called iMet-Q, which provides highly accurate values. In addition, we have proposed a computational method for metabolite identification, which includes an effective clustering step to group a metabolite and its fragments, followed by searches against different metabolite databases. Currently, we are developing an automated tool to implement our methods.
Taiwan Cancer Moonshot Project.
In August 2016, Taiwan was invited by the U.S. National Cancer Institute (NCI) to join the international Cancer Moonshot Project. A major goal that we are working toward is proteogenomic characterization of cancers. We are especially focused on early onset and early stage lung cancer, for which many mutations are similar. One major challenge is to use MS data to detect variant peptides (peptides arising from mutations). In order to do so, we are designing computational methods that are specific for identification of variants as well as developing tools for researchers to choose appropriate proteases for protein digestion. Proper digestion prior to MS experimentation will help to render variant peptides identifiable. Additionally, our new Multi-Q 2 tool will be applied for quantitation of TMT 10-plex labeled samples. We are currently analyzing MS big data acquired from lung cancer patient tissues. According to NCI criteria, we must identify at least 10,000 proteins from each sample to produce a sufficient profile. Along with the basic profiling, we also aim to find variant peptides.
Collaborators
Since bioinformatics is an interdisciplinary research area, we have many collaborations with principal investigators from Agriculture Biotechnology Research Center (ABRC), Biodiversity Research Center (BRC), the Genomics Research Center (GRC), Institute of Biomedical Science (IBMS), Institute of Chemistry (IC), Institute of Cellular and Organismic Biology (ICOB), Institute of Molecular Biology (IMB), Institute of Plant Science and Microbiology (IPSM), and Institute of Statistical Sciences (ISS) at Academia Sinica; National Health Research Institute, Taiwan; and College of Bioresources and Agriculture, College of Life Science, and College of Medicine at National Taiwan University; College of Life Science at National Cheng Kung University; Department of Aquaculture at National Taiwan Ocean University; Fisheries Research Institute; and physicians from National Taiwan University Hospital. Furthermore, we also have international collaborative research projects with the Department of Plant Biology and Medical School at Michigan State University, David Geffen School of Medicine at UCLA, Institute for Protein Research at Osaka University (JAPAN), and National Institute of Advanced Industrial Science and Technology (AIST) in Japan.